Autores
- Paloma Martínez Sebastiá
- Mariana Serejo Soares Branco
- Juan Malo Ascaso
- Laura Cabezuelos Otal
- Lucía Cobano Humanes
- María Dolores Monedero Picazo
- Luis Requeni Monfort
- Hospital Universitario Dr. Peset, Valencia
- Correspondencia: [email protected]
Historia Clínica
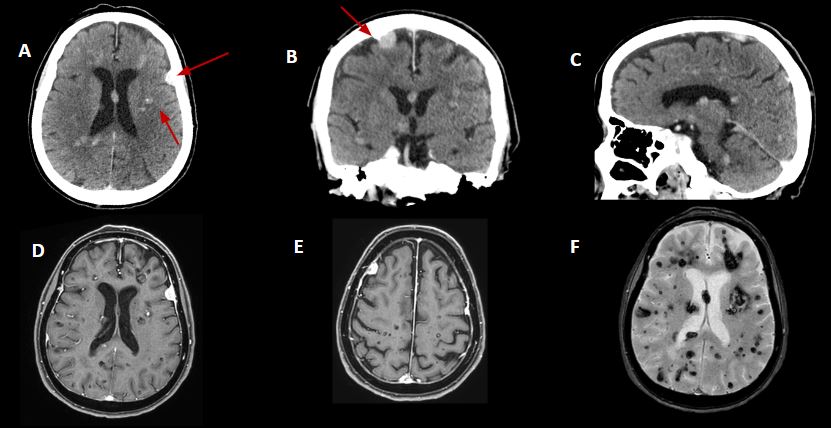
TC de cráneo en plano axial, coronal y sagital con contraste (Imágenes A. B, C). Múltiples lesiones nodulares intraaxiales bilaterales supra e infratentoriales, alguna con calcificaciones internas. Asocian lesiones extraaxiales también bilaterales con realce homogéneo de contraste que sugieren meningiomas.
RM con secuencias potenciades en T1 con contraste (Imágenes D y E) y T2 gradiente (Imágen F). Se confirma la presencia de múltiples lesiones bilaterales con morfologia en “palomita de maíz” en relación con cavernomas múltiples y la presencia de lesiones extraaxiales con realce homogéneo y cola dural compatibles con meningiomas.
Diagnóstico.
Cavernomatosis bilateral asociado a meningiomas bilaterales (PCD10 mutado).
Los meningiomas son la neoplasia intracraneal benigna más común, presentándose como tumores múltiples en menos del 10% de los casos. La NF2 es la afección genética más común que presenta meningiomas múltiples, también descritos en otros síndromes genéticos, incluidos la schwannomatosis, los síndromes de Werner y Proteus.
Las malformaciones cavernosas cerebrales (MCC) representan aproximadamente el 15% de todas las lesiones vasculares cerebrales, con una incidencia familiar de casi el 20%. Se define como síndrome de malformación cavernosa múltiple familiar cuando existe uno o más de los siguientes: cinco o más cavernomas o un cavernoma y al menos otro miembro de la familia con uno o más cavernomas, o mutaciones en uno de los tres genes KRIT1 , CCM2 o PDCD10.
Las MCC se visualizan en RM como lesiones en “palomitas de maíz” con un núcleo de intensidad de señal mixta, generalmente rodeado por un borde hipointenso de hemosiderina en secuencias ponderadas en T2. Las secuencias de imágenes de eco de gradiente (GRE) ponderadas en T2* y de susceptibilidad se consideran las más sensibles para identificar múltiples CCM.
Los meningiomas típicos aparecen como masas extraaxiales de base dural amplia, isointensos a la sustancia gris y que realzan intensamente tras la administración de contraste, con o sin presencia de cola dural.
Existe una asociación en la que todos los individuos genéticamente evaluados con CCM y meningioma presentaban mutaciones en el gen PDCD10 siguiendo una herencia autosómica dominante.
Bibliografía:
Garaci F, Marsili L, Riant F, Marziali S, Cécillon M, Pasquarelli R, Sangiuolo F, Floris R, Novelli G, Tournier-Lasserve E, Brancati F. Cerebral cavernous malformations associated to meningioma: High penetrance in a novel family mutated in the PDCD10 gene. Neuroradiol J. 2015 Jun;28(3):289-93. doi: 10.1177/1971400915591688. PMID: 26246098; PMCID: PMC4757286.
Rosário Marques I, Antunes F, Ferreira N, Grunho M. Malformación cavernosa cerebral familiar: informe de una nueva mutación KRIT1 en una familia portuguesa. Convulsión. 2017;53:72-4.
Mespreuve M, Vanhoenacker F, Lemmerling M. Síndrome de malformación cavernosa múltiple familiar: características de la RM en esta amenaza poco común pero silenciosa. J Belg Soc Radiol. 2016;100(1):51.
Deja una respuesta
Lo siento, debes estar conectado para publicar un comentario.