Autores
- Mª Montserrat Novoa Ferro [email protected]
- Mª Eloísa Santos Armentia
- Noelia Silva Priegue
- Hospital POVISA, Vigo (Pontevedra)
Historia Clínica
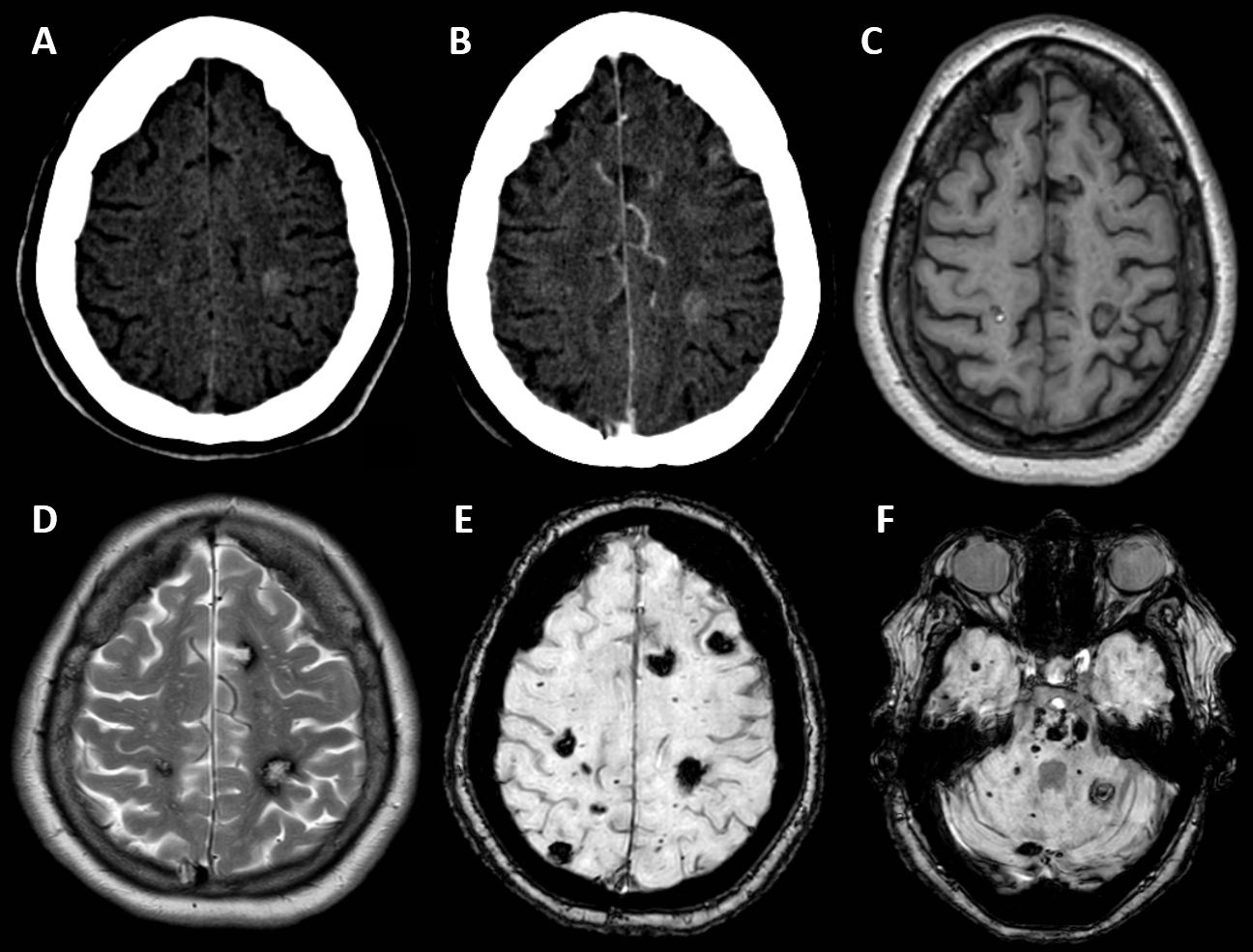
Diagnóstico.
Síndrome de malformación cavernomatosa múltiple familiar con mutación genética KRIT1.
Síndrome de malformación cavernomatosa múltiple familiar con mutación genética KRIT1.
Los cavernomas son malformaciones vasculares en cerebro y médula, constituidas por agrupaciones de vasos inmaduros dilatados sin tejido neural normal interpuesto.
El síndrome de malformación cavernomatosa múltiple familiar únicamente constituye entre el 5-15% de las malformaciones vasculares cerebrales. Aunque puede ser esporádica en 1/3 de los casos, generalmente tiene una herencia autosómica dominante. Las mutaciones genéticas más frecuentemente descritas son las siguientes: KRIT1 (53-65%), CCM2 (20%), PDCD10 (10-16%).
Aunque estas malformaciones se encuentran a cualquier edad, generalmente son asintomáticos (50%) y cuando se manifiestan lo hacen entre la 2ª y 5ª década de la vida, al producirse sangrados intracerebrales que pueden provocar convulsiones, déficits neurológicos focales o cefaleas. Además pueden asociar lesiones vasculares cutáneas (9%) y retinianas (5%).
Para el diagnóstico por imagen, la técnica de elección es la RM donde son de gran utilidad las secuencias de eco de gradiente y susceptibilidad magnética que hacen mucho más visibles los milimétricos focos hipointensos secundarios a residuos de hemosiderina.
Ante múltiples hemorragias cerebrales se debe realizar el diagnóstico diferencial fundamentalmente entre: angiopatía amiloide (edad avanzada, localización corticosubcortical), encefalopatía hipertensiva (antecedente de HTA, localización ganglios de la base y fosa posterior), daño axonal difuso (antecedente traumático, localización unión sustancia blanca-gris) y metástasis hemorrágicas (antecedente neoplásico, realce con contraste).
El manejo de los cavernomas es conservador porque generalmente son asintomáticos, aunque las lesiones que producen síntomas pueden resecarse cuando son accesibles.
BIBLIOGRAFÍA:
Deja una respuesta
Lo siento, debes estar conectado para publicar un comentario.